
1. Introduction¶
1.1. Bayesian Analysis of Macroevolutionary Mixtures¶
BAMM (Bayesian analysis of macroevolutionary mixtures) is a program for modeling complex dynamics of speciation, extinction, and trait evolution on phylogenetic trees. The program is oriented entirely towards detecting and quantifying heterogeneity in evolutionary rates. BAMM uses reversible jump Markov chain Monte Carlo to automatically explore a vast universe of candidate models of lineage diversification and trait evolution. BAMM and associated methods have been described and extended in several publications (PLoS ONE 2014 , Nature Communications 2013 , Systematic Biology 2014, and Evolution 2015). BAMM is a command line program written in C++. Post-run analysis and visualization is performed using the R package BAMMtools.
1.2. Why Use BAMM?¶
Allows modeling of rates through time and among clades. BAMM abandons the simple notion of rate shifts involving constant-rate diversification processes. A rate shift in the BAMM environment is a shift in the evolutionary dynamics of a system and can involve shifts to new time-varying or diversity-dependent evolutionary rate regimes.
Detects evolutionary rate heterogeneity. BAMM allows explicit tests of the hypothesis that a single process of diversification can account for a given phylogenetic pattern. The program automatically explores many candidate models of diversification and provides a variety of tools for interpretation. BAMM enables estimation of Bayesian credible intervals on the number of distinct macroevolutionary regimes, calculation of Bayes factors for pairwise evaluation of model fit, and calculation of model posterior probabilities. The user can easily determine the evidence favoring rate variation within phylogenies.
Detects key innovations and diversity-dependence. BAMM is oriented towards the automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. BAMM finds locations for shifts in evolutionary dynamics that are maximally supported by the data, with no a priori specification as to where these shifts in dynamics might have occurred.
Analysis and visualization with BAMMtools. BAMMtools is a comprehensive R library for the analysis and visualization of macroevolutionary dynamics. The package includes a variety of functions for analyzing and visualizing BAMM output. Examples of BAMMtools functionality includes: visualization of distinct evolutionary regimes on phylogenetic trees, calculation of Bayes factors, plotting evolutionary rates through time, estimating clade-specific average rates, and visualizing rate variation along the branches of individual phylogenetic trees (as shown in this figure).
It is better than stepwise AIC. Many methods for modeling evolutionary dynamics use stepwise AIC-based approaches that are limited to identifying a single best model. These approaches are inherently limited, because many distinct combinations of evolutionary shift regimes might have roughly equal probabilities. Rather than identifying a single best configuration of rate shifts, BAMM samples rate shift configurations in proportion to their posterior probability. Please read our detailed explanation of problems associated with stepwise AIC and other methods that identify a single “best” rate shift configuration.
It is fast. BAMM’s underlying C++ core allows rapid modeling of evolutionary dynamics in phylogenetic datasets that would be too large to handle with R alone.
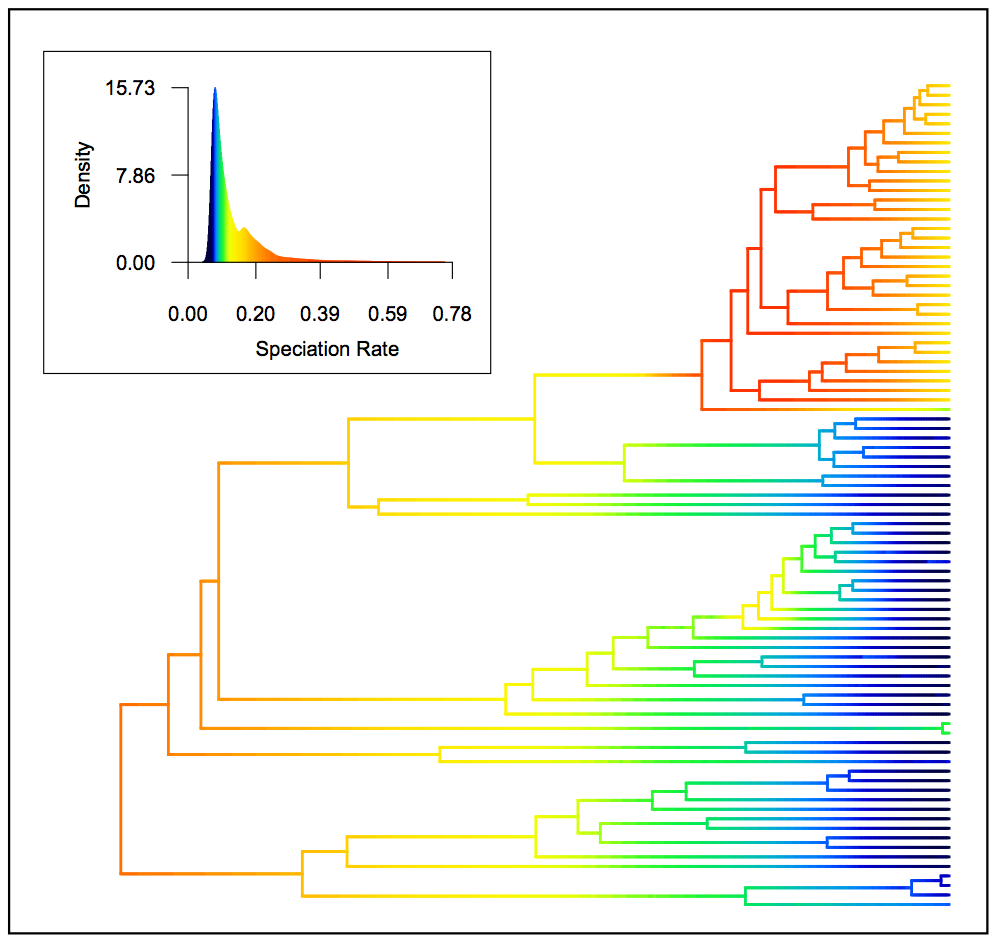
Speciation rates in whales, reconstructed using BAMM. BAMM can estimate the
posterior density of evolutionary rates (speciation, extinction, trait
evolution) at any point in time along any branch in a phylogenetic tree.
Major burst in rates at top of tree corresponds to the origin of the dolphin
clade. See the BAMM graph gallery for more examples involving BAMM. Inset
histogram shows posterior density of speciation rates and allows color-based
interpretation of rates on the whale phylogeny. Figure was generated using
the plot.bammdata(...) function from the BAMMtools package in R. Dataset in
this example is taken from M. Steeman et al., Syst. Biol. 58:573-585,
2009).